IMMER NOCH NICHT OPTIMAL – STUDIEN UND ZULASSUNGSENTSCHEIDUNGEN
Als CONTERGAN 1957 in den Handel kam, war der stark expandierende Arzneimittelmarkt Deutschlands ohne jegliche Kontrolle. Im Gegensatz zu anderen Ländern der Europäischen Wirtschaftsgemeinschaft (EWG) hatte die Bundesrepublik Deutschland kein Arzneimittelgesetz (AMG) – ein beträchtlicher Nachteil für einen Staat, der zur damaligen Zeit den Ruf der „Apotheke der Welt“ hatte.
ZULASSUNG MIT WEBFEHLERN: Als das AMG 1961 in Kraft trat, war es durch die Ereignisse um CONTERGAN mit tausenden Geschädigten (s. a-t 2019; 50: 93-5) bereits überholt. Denn als Basis für die Vermarktung von Arzneimitteln sah es lediglich eine Registrierung beim damaligen Bundesgesundheitsamt (BGA) vor – ohne Prüfung auf Wirksamkeit und Sicherheit. Erst mit dem AMG von 1976 (in Kraft getreten 1978) mussten für die Zulassung Qualität, Wirksamkeit und Unbedenklichkeit nachgewiesen werden. Zu diesem Zeitpunkt waren allerdings dem BGA bereits 140.000 Fertigarzneimittel* angezeigt worden. Eine gigantische Hypothek, denn diese „Alt-Arzneimittel“ mussten nun nachzugelassen werden. Deren Überprüfung auf Wirksamkeit und Unbedenklichkeit sollte einschließlich Abverkaufsfrist bis Ende 1992 abgeschlossen sein.1 Dennoch waren noch 25 Jahre später Alt-Arzneimittel im Handel, die lediglich „fiktiv“ zugelassen waren (a-t 2018; 49: 39).
| * | Dabei wurde jede Packungsgröße gezählt. |
Positive Folge von AMG und Nachzulassung war, dass der Arzneimittelmarkt nicht weiter unkontrolliert wuchs, sondern zunächst sogar etwas schrumpfte. Auf der Strecke blieb beispielsweise eine Vielzahl irrationaler Kombinationen (siehe a-t 2019; 50: 98-100). Anbieter, die bisweilen nicht die erforderlichen Nachweise liefern konnten – oder aus finanziellen Gründen nicht liefern wollten –, wählten, soweit dies möglich war, andere Vermarktungsstrategien: Aus registrierten Alt-Arzneimitteln, beispielsweise Vitamin- oder pflanzlichen Präparaten, wurden Nahrungsergänzungsmittel, für die keine behördliche Zulassung erforderlich ist, oder etwa aus Arzneimitteln zur Befeuchtung der Augen wurden Medizinprodukte.
Die Hoffnung, durch die neuen Anforderungen des AMG unzureichend geprüfte und riskante Arzneimittel vom Markt fernzuhalten, erfüllte sich jedoch nicht. Denn für die Wirksamkeitsbewertung ist im Rahmen der Zulassung lediglich zu klären, ob ein statistisch nachweisbarer Effekt dokumentiert ist, wobei zum Teil auch Studien akzeptiert werden, in denen das Prüfprodukt lediglich mit Plazebo verglichen bzw. auf der Basis von Surrogatkriterien (s. unten) geprüft wird oder nur geringe Patientenzahlen einbezogen sind. Unter diesen Voraussetzungen sind keine zuverlässigen Rückschlüsse auf den tatsächlichen klinischen Nutzen des Prüfpräparates möglich. Nach § 5 des AMG darf zudem die Zulassung noch nicht einmal dann versagt werden, wenn „therapeutische Ergebnisse nur in einer beschränkten Zahl von Fällen erzielt worden sind“.
Das AMG 1976 spiegelt die politische Zielsetzung der 1960er und 1970er Jahre wider, auch den so genannten Besonderen Therapierichtungen unter dem Deckmantel des „Wissenschaftspluralismus“ Zulassungen zu ermöglichen, mit einem Verfahren, bei dem anstelle der eigentlich erforderlichen aussagekräftigen Studien anderes Erkenntnismaterial ausreicht, beispielsweise Expertenurteile von Sachverständigen (z.B. Kommission D für Homöopathie** oder E für Phytotherapeutika), die über wissenschaftliche Kenntnisse und Erfahrungen mit der jeweiligen Therapierichtung verfügen. Mit diesem so genannten Binnenkonsens wird umgangen, dass eine kritische Bewertung der Datenlage durch unabhängige Datenerhebungen erfolgen muss (a-t 2019; 50: 49-50). Somit sagt eine Zulassung der „Besonderen Therapierichtungen“ nichts über einen Wirksamkeitsnachweis aus.
| ** | Homöopathika ohne Indikationsangaben werden bis heute nicht zugelassen, sondern lediglich registriert. |
Insbesondere Anbieter umstrittener Arzneimittel betonen hingegen bisweilen, dass die Zulassung eine Art Qualitätssiegel sei. Entsprechend warb die Firma Bionorica Ende der 1990er Jahre unter Bezug auf die Neuzulassung von SINUPRET FORTE, das fünf pulverisierte pflanzliche Bestandteile enthält und bei Nasennebenhöhlenentzündung angewendet werden soll, mit „dokumentierter Qualität und Wirksamkeit“. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) verwies jedoch auf unsere Frage nach der Grundlage der Zulassung lediglich auf einen Monografie-Entwurf der Kommission E (a-t 1998; Nr. 4: 37-8).
Bis 1995 erfolgten die Zulassungen*** in Europa durch nationale Behörden, in Deutschland durch das BGA bzw. seit 1994 durch das BfArM oder das für Impfstoffe und Sera sowie später auch für biologisch hergestellte Arzneimittel zuständige Paul-Ehrlich-Institut (PEI). Seither ermöglicht das EU-Recht neben der zentralen Zulassung bei der europäischen Arzneimittelbehörde EMEA, heute EMA, die für bestimmte Arzneimittel wie Biologika und Orphan Drugs verpflichtend ist, auch eine nationale Zulassung, die dann auf weitere Mitgliedsstaaten ausgedehnt werden kann (Verfahren der gegenseitigen Anerkennung).
| *** | Tierarzneimittel werden vom Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (BVL) zugelassen. |
Die verschiedenen Verfahren lassen grundsätzlich keine Rückschlüsse auf die Qualität der Zulassungen zu. Das europäische Verfahren ist allerdings insofern transparenter, als die EMA routinemäßig ausführliche öffentliche Bewertungsberichte (EPAR; European Public Assessment Report) im Internet veröffentlicht. Entsprechende Unterlagen stehen vom BfArM und PEI sowie von den anderen Behörden der EU-Länder zumeist nicht zur Verfügung.
Beim Vergleich der Tätigkeit von Zulassungsbehörden international steht oft die Geschwindigkeit der Zulassungen mehr im Blickpunkt als die Qualität (a-t 1998; Nr. 4: 37-8). Pharmahersteller und von diesen gesponserte Patientengruppierungen fordern bisweilen von den Behörden, die Zulassung von Arzneimitteln zu beschleunigen. Gängiges Argument ist, dass so die Patienten rascher mit neuen Arzneimitteln behandelt werden können. Damit wird suggeriert, dass neue Arzneimittel nahezu automatisch patientenrelevante Vorteile bringen. Dass dies nicht die Regel ist, ist eine häufige Erfahrung, wenn das a-t Neueinführungen bewertet – z.B. bei Andexanet alfa (ONDEXXYA, a-t 2019; 50: 74 und e a-t 9/2019), Buprenorphin-Depot (BUVIDAL; a-t 2019; 50: 36-8) oder Dapagliflozin (FORXIGA; a-t 2019; 50: 57-9) bei Typ-1-Diabetes. Wichtiger als Geschwindigkeit ist unseres Erachtens, Patienten zuverlässig vor unzureichend geprüften Arzneimitteln zu schützen, die sich nicht oder nur schlecht hinsichtlich Nutzen und Schaden in die vorhandenen Therapieoptionen einordnen lassen. Profiteure einer beschleunigten Zulassung sind nur selten die Patienten, aber immer die Pharmaanbieter, die ihre Produkte rascher vermarkten und so die wirtschaftlich lukrative patentgeschützte Vermarktungsphase ihrer Präparate verlängern können (a-t 2015; 46: 119-21).
FRÜHE NUTZENBEWERTUNG: Um eine Einordnung des Nutzens neuer Arzneimittel gegenüber etablierten Therapien zu ermöglichen, durchlaufen sie in Deutschland seit 2011 nach dem Arzneimittelmarktneuordnungsgesetz (AMNOG) bei Markteinführung und Indikationserweiterung eine frühe Nutzenbewertung. Für die wissenschaftliche Beurteilung wird in der Regel das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) beauftragt. Für die abschließende Bewertung ist der Gemeinsame Bundesausschuss (G-BA; s. auch Seite 107) zuständig.****
| **** | Der G-BA besteht aus Vertretern des GKV-Spitzenverbandes, der Deutschen Krankenhausgesellschaft (DKG), der Kassenärztlichen und Kassenzahnärztlichen Bundesvereinigung (KBV, KZBV) sowie unparteiischen Mitgliedern, von denen einer den Vorsitz stellt. |
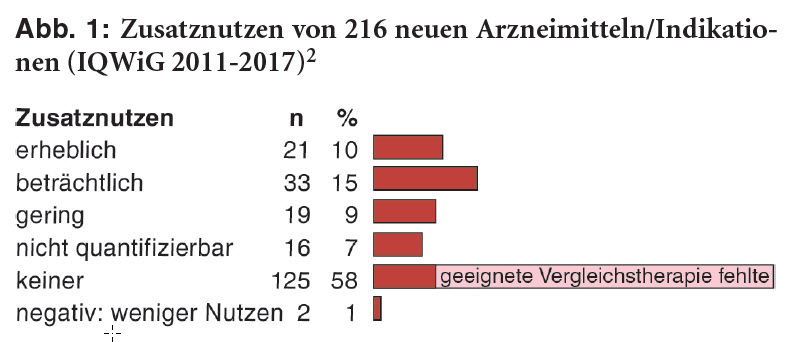
Eine aktuelle Analyse der IQWiG-Beurteilungen der 216 von 2011 bis 2017 bewerteten Arzneimittel (152 neue Wirkstoffe, 64 neue Indikationen) ernüchtert: Nur bei 25% wird ein beträchtlicher oder erheblicher Zusatznutzen festgestellt und bei 16% ein geringer oder nicht quantifizierbarer. Bei rund 40% der Mittel mit Zusatznutzen bezieht sich dieser zudem nur auf Teilbereiche der zugelassenen Indikationen. Zwei Arzneimittel (1%) sind schlechter als Standardbehandlungen. Für mehr als die Hälfte (125; 58%) kann dagegen kein Zusatznutzen attestiert werden. Dies beruht aber lediglich bei 9% der Arzneimittel (n = 19) tatsächlich auf Abgleichen mit einer vom G-BA vorgegebenen zweckmäßigen Vergleichstherapie (s. Abbildung 1). Bei den übrigen Arzneimitteln ohne Zusatznutzen (106; 49%) ist eine Bewertung überhaupt nicht möglich, weil nur Vergleichsstudien mit Plazebo (n = 64) oder Therapien vorlagen, die vom G-BA nicht als geeignet angesehen waren (n = 42).2 Die IQWiG-Bewertungen können somit insgesamt keinen aussagekräftigen Überblick über den möglihen therapeutischen Nutzen der im Rahmen der frühen Nutzenbewertung überprüften neuen Arzneimittel geben.
STUDIEN UNTER EINFLUSS: Da öffentliches Geld knapp ist, werden Studien hauptsächlich von Arzneimittelfirmen finanziert. Dies wirkt sich auf die Themenwahl und auch auf die Konzeption der Untersuchungen aus, die Ergebnisse begünstigen können, die für den Anbieter des Prüfpräparates positiv sind (a-t 2010; 41: 1-3). Arzneimittelhersteller sind nicht dazu verpflichtet, Forschung am Bedarf des Allgemeinwohls auszurichten. Firmenmanager stehen jedoch unter dem Druck der Anteilseigner, den Gewinn bzw. den Wert der Aktien zu steigern. Sie finanzieren daher vorwiegend Studien, die Firmeninteressen nachgehen und nicht der Klärung therapeutisch relevanter Fragen. Nach einer 2003 publizierten systematischen Übersicht gehen klinische Studien, Metaanalysen klinischer Studien und pharmakoökonomische Untersuchungen, die von Pharmaherstellern gesponsert werden, viermal so häufig zu Gunsten des Prüfpräparates aus wie Studien mit anderen Geldgebern. Diese systematische Verzerrung (Bias) zu Gunsten firmeneigener Produkte kommt beispielsweise durch Auswahl ungeeigneter oder fehldosierter Vergleichspräparate zu Stande. Zudem werden firmenfinanzierte Studien demnach seltener publiziert als Untersuchungen anderer Sponsoren und häufiger auf Kongressen vorgestellt (Publikationsbias). Dort ist wegen des fehlenden Peer-Reviews eine für das Produkt günstige Ergebnisinterpretation möglich, selbst wenn diese durch die Daten nicht gedeckt ist (a-t 2003: 34: 62-3). Eine Nachfolgeübersicht, die mehr als 2.600 Studien der Jahre 2003 bis 2006 einbezieht, geht mit diesem Ergebnis konform (a-t 2010; 41: 1-3).
SURROGATPARAMETER: Klinische Studien sollten genügend groß und ausreichend lang angelegt sein und das Prüfpräparat randomisiert bezüglich patientenrelevanter Endpunkte möglichst mit der aktuellen Standardtherapie vergleichen. Surrogatparameter erweisen sich hingegen zur Beurteilung des klinischen Nutzens oft als irrelevant und manchmal sogar als irreführend. Dies hat sich zum Beispiel an der Langzeitprävention mit Sexualhormonen nach den Wechseljahren gezeigt, die sich jahrzehntelang praktisch ausschließlich auf Surrogatkriterien wie günstig beeinflusste Blutwerte sowie Beobachtungsstudien stützte – bis randomisierte Interventionsstudien mit patientenrelevanten Endpunkten eine negative Nutzen-Schaden-Bilanz belegten (a-t 2002; 33: 81-3).
Von 36 Onkologika, die die FDA zwischen 2008 und 2012 auf der Basis von Surrogatparametern wie Ansprechrate oder progressionsfreies Überleben zugelassen hat, findet sich bei 31 (86%) nach im Median 4,4 Jahren weiterhin kein Nachweis eines positiven Effektes auf das Gesamtüberleben. Dieser ließ sich lediglich für 5 Mittel (14%) bestätigen (a-t 2016; 47: 14-5). Ebenfalls enttäuschend fällt eine entsprechende Auswertung von Onkologika aus, die unter EMA-Zuständigkeit zwischen 2009 und 2013 auf Basis von Surrogatkriterien zugelassen worden sind: Nach im Median 5,4 Jahren findet sich ein Überlebensvorteil lediglich bei 26 (38%) von 68 Indikationen und eine verbesserte Lebensqualität bei 18%, soweit diese überhaupt mituntersucht worden ist ( a-t 2017; 48: 104-5).
Arzneimittel, die auf der Basis von Surrogatkriterien zugelassen worden sind, werden üblicherweise nicht aus dem Handel gezogen, wenn nach der Zulassung der Nachweis eines patientenrelevanten Nutzens ausbleibt. Eine Ausnahme ist Olaratumab (LARTRUVO). Der Hemmer des Thrombozytenwachstumsfaktor-Rezeptors alpha wurde aufgrund eines Hinweises auf eine Verlängerung des Überlebens aus einer darauf primär nicht angelegten offenen Phase-II-Studie unter Auflagen beschleunigt zugelassen, später aber wegen Unwirksamkeit in einer doppelblinden Phase-III-Studie zurückgezogen. Ein gesetzlich geregelter Entschädigungsanspruch der Krankenkassen, die knapp 40 Mio. € für den Antikörper ausgegeben haben, fehlt bislang (a-t 2019; 50: 55).
MANIPULIERTE STUDIENDATEN: Eine Grundvoraussetzung für die Bewertung von Arzneimitteln ist, dass alle klinischen Studien tatsächlich veröffentlicht und somit zugänglich sind – unabhängig davon, ob diese für das Prüfpräparat positiv oder negativ ausfallen. Nach einer 2013 publizierten Auswertung von 585 in der US-amerikanischen Studiendatenbank ClinicalTrials.gov erfassten und vor Januar 2009 abgeschlossenen Studien war jede dritte bis vierte im Median fünf Jahre nach Studienabschluss immer noch nicht veröffentlicht (e a-t 11/2013). Wenn Studienpublikationen ausbleiben, kann dies eine gezielte Unterdrückung von Informationen sein, die aus Firmensicht unerwünscht sind:
So versuchte Pfizer 2010 das IQWiG mit einer Vorauswahl von Studien für Reboxetin (EDRONAX) abzuspeisen und wollte zwei Drittel aller bislang zu dem Antidepressivum in Studien erhobenen klinischen Daten unter Verschluss halten. Erst unter öffentlichem Druck gab der Konzern die Blockade auf. Der Hintergrund war klar: Die publizierten Studien suggerierten einen Nutzen von Reboxetin, die Gesamtschau der Daten lässt hingegen keinen Nutzen erkennen (a-t 2010; 41: 1-3 und 111-2). Das Antidepressivum ist daher seit 2010 nicht mehr zu Lasten der GKV verordnungsfähig und seit 2019 außer Handel.
Der Neuraminidasehemmer Oseltamivir (TAMIFLU) soll nach einer 2003 publizierten gepoolten Auswertung von zehn randomisierten Studien schwere Komplikationen einer Virusgrippe deutlich reduzieren. Die Studie stammt allerdings überwiegend von Angestellten und bezahlten Beratern des Anbieters Roche, und acht der zehn Studien waren nicht vollständig veröffentlicht (a-t 2010; 41: 4, 13-4). Noch 2011 – neun Jahre nach der Markteinführung – waren die Daten von 60% der Patienten aus Phase-III-Studien zur Influenzatherapie mit Oseltamivir nicht veröffentlicht (a-t 2012; 43: 17-8). Erst 2014 konnten Cochrane-Autoren – wiederum nach öffentlichem Druck – die zuvor von Roche zurückgehaltenen unveröffentlichten Zulassungsstudien berücksichtigen. Unter Einschluss dieser Daten bleibt der Beleg aus, dass Oseltamivir das Risiko von Komplikationen einer Influenza wie Pneumonie, Klinikeinweisung oder Tod reduzieren kann. Für Roche hat sich das Zurückhalten relevanter Studiendaten ausgezahlt. Oseltamivir wurde zum Goldesel: Die Bundesregierung hatte den Neuraminidasehemmer nach der Formel „zugelassen, also wirksam“ und auf Anraten des Robert Koch-Instituts trotz vielfach geäußerter Vorbehalte (a-t 2005; 36: 62-3, 2006; 37: 22-3, 2009; 40: 77-80 sowie 2010; 41: 69-70) für etwa 500 Mio. Euro eingelagert. 2009 hatten 96 Staaten so viel Oseltamivir eingekauft, dass geschätzt 350 Millionen Menschen damit hätten behandelt werden können (a-t 2014; 45: 54-5).
Wesentliche Voraussetzung für die Bewertung von Arzneimitteln ist auch, dass die Studien wissenschaftlich korrekt durchgeführt und vollständig publiziert werden. Die Manipulationsmöglichkeiten, um Ergebnisse zu schönen, sind allerdings vielfältig und der Fantasie offensichtlich keine Grenzen gesetzt. Wer würde auf die Idee kommen, dass den Zulassungsbehörden andere Studienfassungen eingereicht als später – wenn überhaupt – veröffentlicht werden? FDA-Mitarbeiter haben 74 Studien zu zwölf Antidepressiva nachverfolgt, die bei der FDA zwischen 1984 und 2004 eingereicht wurden und zur Hälfte positiv (51%; n = 38) bzw. negativ oder nicht eindeutig (49%; n = 36) ausgefallen waren. Veröffentlicht fanden sie davon anschließend lediglich 51 der ursprünglich 74 Studien wieder, von denen aber jetzt 48 (94%) das geprüfte Antidepressivum positiv beschreiben und nur 3 (6%) negativ. Die Aufbesserung der Datenlage für die Psychopharmaka beruht also darauf, dass zahlreiche Negativstudien nicht veröffentlicht und andere so manipuliert worden sind, dass sie ein positives Ergebnis vortäuschen (a-t 2010; 41: 1-3). Schwedische Autoren, die der schwedischen Behörde eingereichte Studien zu selektiven Serotonin-Wiederaufnahmehemmern (SSRI) mit den tatsächlich publizierten Daten verglichen haben, kamen zuvor bereits zu ähnlichen Ergebnissen. Sie folgerten, dass jegliche Empfehlung eines SSRI auf der Basis veröffentlichter Daten auf verzerrter Datenlage beruht (a-t 2003; 34: 62-3). Aber selbst wenn gravierende Manipulationen erkannt sind, werden die Studien in aller Regel nicht zurückgezogen oder korrigiert (a-t 2011; 42: 25-6).
Manipulationen und Nichtveröffentlichung negativer Daten (Publikationsbias) erachten wir als ethisch zutiefst verwerflich. Sie bedeuten eine grobe Missachtung der Bereitschaft der Patienten, die Unabwägbarkeiten einer Studie für den wissenschaftlichen Erkenntnisgewinn auf sich zu nehmen. Die Manipulationen haben zudem langfristige Folgen. Sie wirken sich auch auf Metaanalysen aus, die überwiegend veröffentlichte Daten einbeziehen, sowie auf Leitlinien, die auf den Ergebnissen von Studien und Metaanalysen basieren. Falsche und wissenschaftlich nicht begründbare Therapien sind die Folge.
ZULASSUNG AUF UMWEGEN: Lehnen Behörden die Zulassung eines Medikamentes ab, nutzen manche Anbieter Schlupflöcher in der Arzneimittelgesetzgebung oder gehen juristisch durch alle gerichtlichen Instanzen, um ihr Produkt dennoch auf den Markt zu bringen:
So versagte das BfArM zwar die Nachzulassung für das zur Therapie von Gelenkerkrankungen propagierte Oxaceprol (AHP 200) wegen unzureichender Nutzenbelege. Nach Beschwerden des Anbieters Rosen Pharma gewährte schließlich das Bundesverwaltungsgericht die Zulassung auf der Basis eines rumänischen Oxaceprol-Präparates, dessen Indikation noch nicht einmal mit der zunächst für AHP beanspruchten übereinstimmte. Aussagekräftige Studien fehlen jedoch nach wie vor (a-t 2018; 49: 47).
2015 nahm die Firma Strathmann Pridinol (MYOSON DIRECT) aus dem Handel, weil die Studiendaten für eine Nachzulassung nicht ausreichten. Zwei Jahre später erhielt sie eine Neuzulassung des vermeintlichen Muskelrelaxans (jetzt als MYOPRIDIN), die lediglich auf Bioäquivalenz mit einem bereits seit 1961 in Italien zugelassenen Präparat beruht. Auf unsere Fragen nach Studien für den Nutzenbeleg hat es die Firma vorgezogen, nicht zu antworten (a-t 2018; 49: 45).
AUSBLICK: Zulassungen sollten nach gründlicher Prüfung ohne Zeitdruck erfolgen. Für Arzneimittel zur Langzeitmedikation sind zwingend Langzeitstudien erforderlich. Surrogatkriterien, etwa Senkung des Cholesterinspiegels oder des Blutdrucks, reichen nicht aus, um die Wirksamkeit eines Arzneimittels zuverlässig beurteilen zu können (a-t 2001; 32: 88-9).
Zulassungsentscheidungen müssen nachvollziehbar sein. Deshalb sollten die entscheidungsrelevanten klinischen Studien spätestens zum Zeitpunkt der Zulassung öffentlich zugänglich sein. Sollte eine Zulassung nur unter Auflage einer Sicherheitsstudie oder einer ergänzenden Studie zum Beleg des Nutzens erfolgen, muss ein definitiver Zeitpunkt angeordnet werden, bis zu dem die entsprechenden Daten der Behörde vorgelegt und öffentlich zugänglich gemacht werden müssen.
| 1 | B-GA: Nachzulassung und -registrierung von 63.000 Alt-Arzneimitteln beim B-GA. Pressemitteilung vom 17. Mai 1990 |
| 2 | WIESELER, B. et al.: BMJ 2019; 366: 14340, online publ. am 10. Juli 2019 (8 Seiten) und 14837, online publ. am 24. Juli 2019 (1 Seite) |
© 2019 arznei-telegramm, publiziert am 15. November 2019
Autor: Redaktion arznei-telegramm - Wer wir sind und wie wir arbeiten
Diese Publikation ist urheberrechtlich geschützt. Vervielfältigung sowie Einspeicherung und Verarbeitung in elektronischen Systemen ist nur mit Genehmigung des arznei-telegramm® gestattet.
