BEISPIEL REBOXETIN (EDRONAX, SOLVEX) Publikationsbias führt Ärzte in die Irre
Vor einem Jahr hat das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) eine Bewertung des Antidepressivums Reboxetin (EDRONAX, SOLVEX) veröffentlicht, nach der ein Nutzen des selektiven Noradrenalin-Wiederaufnahmehemmers nicht nachgewiesen ist, es aber Belege für einen Schaden gibt. Im Vorbericht hatte das Institut noch gänzlich auf eine Auswertung der bis dahin öffentlich zugänglichen Studiendaten verzichtet, da offenkundig war, dass der Hersteller Pfizer zwei Drittel aller bislang in Studien erhobenen Daten unter Verschluss gehalten hatte.1 Erst nach öffentlichem Druck gab die Firma die Blockade auf (a-t 2010; 41: 1-3). Inzwischen hat der Gemeinsame Bundesausschuss Reboxetin von der Verordnungsfähigkeit zu Lasten der Gesetzlichen Krankenversicherung ausgeschlossen. Der Beschluss ist allerdings noch nicht rechtskräftig.2
Jetzt wird ein Teil der Analyse des IQWiG im britischen Ärzteblatt publiziert.3 Neben der metaanalytischen Auswertung veröffentlichter und bislang unveröffentlichter Studien, in denen Reboxetin mit Plazebo und/oder einem selektiven Serotonin-Wiederaufnahmehemmer (SSRI) in der Akuttherapie einer Depression verglichen wird, errechnen die Autoren das Ausmaß der durch die Datenunterdrückung verursachten Verzerrung. In die Analyse gehen 13 Studien mit insgesamt 4.098 Patienten ein. Die Daten von fast 75% der Patienten sind nicht publiziert. Bis auf eine vom Citalopram (CIPRAMIL, Generika)-Anbieter Lundbeck finanzierte Untersuchung sind alle vom Reboxetin-Hersteller gesponsert.3
Die publizierten Studien legen einen Nutzen von Reboxetin im Vergleich zu Plazebo nahe. Werden zusätzlich die unveröffentlichten Studien berücksichtigt, lässt sich jedoch gegenüber Scheinmedikament kein signifikanter Einfluss von Reboxetin auf die Rate der Remissionen* und Responder* mehr nachweisen. Die Rate der Patienten, bei denen mindestens eine unerwünschte Wirkung auftritt oder die die Behandlung wegen Störeffekten abbrechen, ist unter dem Noradrenalin-Wiederaufnahmehemmer hingegen deutlich höher (Odds Ratio [OR] 2,14; 95% Konfidenzintervall [CI] 1,59-2,88 bzw. OR 2,21; 95% CI 1,45-3,37). Dabei sind die positiven Effekte in den veröffentlichten Studien doppelt so groß wie im gesamten Datenpool, das Risiko eines Therapieabbruchs jedoch nur halb so hoch.3
Im Vergleich zu SSRI wirkt Reboxetin bei Analyse aller Daten signifikant schlechter (Remission: OR 0,80; 95% CI 0,67-0,96, Response: OR 0,80; 95% CI 0,67-0,95). Die publizierten Studien, deren Analyse einen etwa gleich großen Nutzen von Reboxetin und SSRI ergibt, überschätzen den Effekt gegenüber dem gesamten Datenpool um rund 20%. Die Auswertung zur Rate der Patienten mit Störwirkungen verändert sich durch die breitere Datenbasis nicht. Der Unterschied bei den Therapieabbrüchen wegen unerwünschter Effekte, die in den veröffentlichten Studien unter Reboxetin numerisch häufiger vorkommen als unter Fluoxetin (FLUCTIN, Generika), ist nun signifikant, am ehesten aufgrund der größeren Power.3
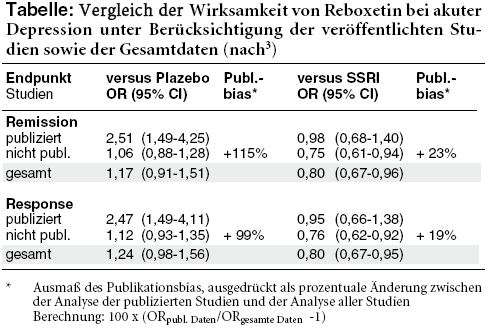
Bei Antidepressiva ist das Problem des selektiven Veröffentlichens besonders ausgeprägt und in verschiedenen Übersichten (z.B. 4,5) gut dokumentiert (a-t 2008; 39: 22; 2003; 34: 62-3). Umso wichtiger ist es, endlich auch in Europa eine Publikationspflicht einzuführen. In den USA müssen seit 2007 die Protokolle und seit 2008 die Ergebnisse aller klinischen Studien (außer Phase-I-Studien) zu US-amerikanischen Arzneimitteln im Studienregister ClinicalTrials.gov in standardisierter Form öffentlich zugänglich gemacht werden. Betreffen die Studien zugelassene Indikationen, sind die Ergebnisse innerhalb eines Jahres nach Abschluss der Untersuchung zu veröffentlichen, bei nicht zugelassenen Indikationen spätestens nach zwei Jahren. Bei Verstößen drohen empfindliche Geldstrafen. Schlupflöcher, die die US-amerikanische Regelung aufweist, zum Beispiel keine Publikationspflicht bei dort nicht zugelassenen Mitteln oder wenn eine Studie vor September 2007 abgeschlossen wurde, gilt es zu schließen.6,7
Die europäische Kommission bereitet zwar seit Längerem einen öffentlich verfügbaren Zugang zu Daten und Ergebnissen aus klinischen Arzneimittelprüfungen vor. Zeitpunkt der Einführung sowie Umfang und Form der zu veröffentlichenden Berichte sind jedoch nach wie vor unklar. Nach dem Willen des deutschen Bundesministeriums für Gesundheit sollen Hersteller unabhängig von den europäischen Plänen die Ergebnisse der von ihnen durchgeführten klinischen Prüfungen zusätzlich selbst öffentlich zugänglich machen. Im Entwurf des Arzneimittelneuordnungsgesetzes (AMNOG)8 ist die Einführung einer Publikationspflicht vorgesehen, die jedoch in erster Linie für Phase III-Studien gelten soll und nur ausnahmsweise für Prüfungen der Phase II oder IV. Eine bestimmte Form der Berichte ist nicht vorgesehen, ebenso wenig ein zentraler Publikationsort. Stattdessen sollen die Daten beispielsweise auf den Webseiten der Firmen bereitgestellt werden8 - unseres Erachtens kein neutraler Weg, um Ärzte und Patienten zu informieren, der zudem die Auffindbarkeit erschwert. Außerdem soll der "Schutz geistigen Eigentums" sowie "der Schutz von Betriebs- und Geschäftsgeheimnissen" gewahrt werden, sodass "im Konfliktfall" eine Veröffentlichung unterbleiben kann.8
Neben einer verbesserten weltweit anzuwendenden Publikationspflicht und dem öffentlichen Zugang zu Studiendaten, die den Zulassungsbehörden vorliegen, fordern die Mitarbeiter des IQWiG in einem begleitenden Kommentar eine bessere Zusammenarbeit zwischen den Behörden.7 Denn nachdem Reboxetin 1997 in mehreren europäischen Ländern zugelassen worden war, ließ die US-amerikanische Arzneimittelbehörde FDA das Mittel zwar zunächst vorläufig zu, lehnte eine endgültige Zulassung jedoch 2001 nach Erhalt weiterer Studiendaten wegen mangelnder Nutzenbelege ab. In Europa blieben die Ergebnisse der neueren Studien und die Entscheidung der FDA ohne Folgen. Abhilfe könnte eine Regelung schaffen, nach der Behörden ein Mittel erneut beurteilen müssen, wenn dessen Zulassung in einem anderen Land versagt wurde.7
Das Beispiel Reboxetin (EDRONAX, SOLVEX) belegt eindrücklich, dass sich Nutzen und Schaden eines Arzneimittels nur dann realistisch einschätzen lassen, wenn alle Studiendaten verfügbar sind.
Eine gesetzliche Regelung einer Publikationspflicht für klinische Studien nach US-amerikanischem Vorbild ist hierzulande überfällig.
| (M = Metaanalyse) | |
| 1 | IQWiG: Pressemitteilung vom 24. Nov. 2009 http://www.iqwig.de/index.981.html |
| 2 | G-BA: Pressemitteilung Nr. 28/2010 vom 16. Sept. 2010 http://www.g-ba.de/downloads/34-215-352/28-2010-09-16-Reboxetin.pdf |
| M 3 | EYDING, D. et al.: BMJ 2010; 341: c4737 (14 Seiten) |
| 4 | MELANDER, H. et al.: BMJ 2003; 326: 1171-5 |
| 5 | TURNER, E.H. et al.: N. Engl. J. Med. 2008; 358: 252-60 |
| 6 | WOOD, A.J.J.: N. Engl. J. Med. 2009; 360: 824-30 |
| 7 | WIESELER, B. et al.: BMJ 2010; 341: c4942 (2 Seiten) |
| 8 | BMG: Entwurf eines Gesetzes zur Neuordnung des Arzneimittelmarktes in der gesetzlichen Krankenversicherung, 29. Juni 2010 http://www.bmg.bund.de/cln_178/nn_1168248/SharedDocs/Downloads/DE/Standardartikel/G/Glossar-Gesetze/amnog,templateId=raw,property=publicationFile.pdf/amnog.pdf |
| * | Abnahme des Punktwerts auf der HAMILTON-Skala überwiegend auf ≤ 10 Punkte (Remission) bzw. um mindestens 50% gegenüber dem Ausgangswert (Response). |
© 2010 arznei-telegramm, publiziert am 5. November 2010
Autor: Redaktion arznei-telegramm - Wer wir sind und wie wir arbeiten
Diese Publikation ist urheberrechtlich geschützt. Vervielfältigung sowie Einspeicherung und Verarbeitung in elektronischen Systemen ist nur mit Genehmigung des arznei-telegramm® gestattet.
